112年3月30日新生兒篩檢暨SMA疾病認知調查發布記者會

- 台灣嬰兒死亡率超日韓兩倍 守護寶寶是關鍵!
- 新生兒篩檢每年篩出三千名健康異常寶寶 但逾一萬五家庭未進行加選篩檢
- 六大學協會籲愈早篩檢與治療、預後愈好
台灣生育率全球最低,連三年負增長,怎麼守護出生寶寶健康是家長常關注的重點!兒童節前夕,國內六大學協會攜手三大篩檢中心,包括臺灣兒科醫學會、台灣小兒神經醫學會、台灣周產期醫學會、台灣新生兒科醫學會、中華民國人類遺傳學會、台灣脊髓肌肉萎縮症病友協會、國立台灣大學醫學院附設醫院新生兒篩檢中心、台北病理中心臨床病理部新生兒篩檢組、中華民國衛生保健基金會附設醫事檢驗所新生兒篩檢中心,共同呼籲及早進行第一道健康檢查—新生兒必選及加選篩檢,包含全世界嬰兒死亡率最高的遺傳性疾病[iii]—脊髓性肌肉萎縮症(SMA)檢測,及早發現問題與治療。
- 為寶寶保險的家長中五成延後篩檢 影響揪疾病根源時間
台灣2022年新生兒數達62年來新低,不僅生得少,資料更顯示每千位嬰兒就有4.1位死亡,高出許多經濟合作暨發展組織(OECD)國家,更已連二十年超過日、韓兩至三倍。
台大醫院基因醫學部主任暨小兒部主治醫師簡穎秀部主任表示,寶寶健康狀況是家長最關心話題,但大多數遺傳性或先天異常疾病在寶寶出生時不會有明顯症狀,因此需要透過新生兒篩檢來把關,且所篩檢出來的疾病可透過藥物或飲食控制,及早降低疾病對身體或智力的損害,能有更好的預後。
新生兒篩檢每年診斷出超過三千名異常個案數,但2023年「新生兒篩檢暨脊髓肌肉萎縮症(SMA)疾病認知調查」卻指出,不少家長為了守護寶寶的未來選擇犧牲現在。在願意讓寶寶進行加選篩檢家長中,超過九成五家長會為寶寶保險,但卻近五成會延後篩檢時間,恐成寶寶健康的隱憂。調查中更發現,晚於常規時間才進行新生兒必選篩檢且為寶寶購買保險的家長中,卻有五成不清楚金融監督管理委員會在民國101年已頒布「新生兒篩檢之政府指定執行21項檢驗項目,皆排除於保險等待期的規範」。簡穎秀部主任提醒,別因商業保險迷思錯失拯救寶寶的機會!
- 出生48小時第一道健康把關—新生兒篩檢
- 三大延誤篩檢主因 恐錯失守護寶寶第一時機!
新生兒篩檢應在甚麼時間點進行?簡穎秀部主任表示,新生兒出生48小時即可篩檢,採取一次足跟血就可以進行必選及加選檢查。目前衛福部國健署有補助新生兒必選篩檢項目,包含先天性甲狀腺低能症、苯酮尿症、楓糖尿症、半乳糖血症等21個項目,也可以額外加選檢測約10項新生兒罕見疾病,為寶寶健康雙重把關。但調查顯示,家長愈晚了解「新生兒篩檢」資訊,對於篩檢意願也愈低,對比現況卻有超過三成家長懷孕晚期才知道相關資訊,令人擔憂。
簡穎秀部主任說,家長一般會在生產後48小時內收到新生兒篩檢的勾選表,決策時間短,因此建議要提早了解。而篩檢時間也很重要,因為先天性代謝異常疾病一出生就會對寶寶造成傷害,及時篩檢有機會及早治療。但觀察願意進行篩檢的家長中,高達五成晚於常規時間才進行篩檢。進一步觀察原因,前三名分別為:決策進行篩檢時間太短、篩檢衛教資訊太少、太晚才開始了解篩檢項目,更有兩成五家長表示已進行產前帶因者檢驗或產前胎兒診斷,認為寶寶很健康。他補充,帶因檢驗並非100%準確,而產後新生兒篩檢更能直接確認寶寶的健康。
- 八成罕病為基因遺傳致 加選篩檢守護寶寶健康!
以2022年出生率數字推估,高達一萬五家庭未進行加選篩檢!進一步觀察不進行「新生兒加選篩檢」原因,三成四認為21項必選篩檢已足夠、近三成認為費用昂貴、近三成認為決策時間太短!台灣小兒神經醫學會秘書長、部立雙和醫院小兒神經科主任郭雲鼎秘書長提醒,罕見疾病當中,高達八成為基因遺傳導致。新生兒加選篩檢涵蓋多種可治療的罕病,如脊髓性肌肉萎縮症(SMA)、黏多醣症(MPS)、龐貝氏症(PD)等疾病檢測。以發生率高的脊髓性肌肉萎縮症(SMA)為例,篩檢準確度高。若延遲篩檢或未篩檢,恐錯過黃金治療時機。
- SMA治療方式多元 愈早篩檢與治療 預後愈好!
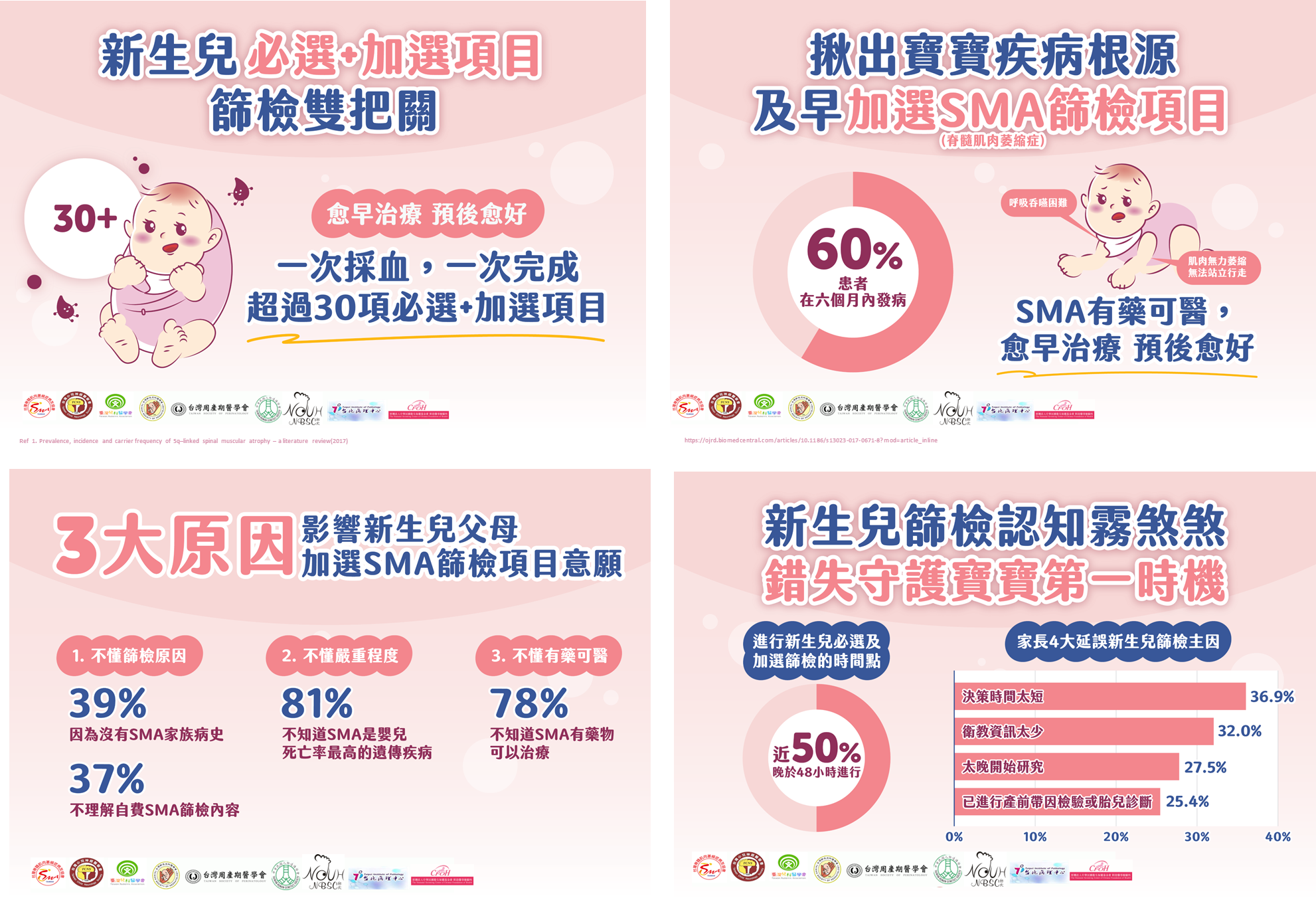
台灣脊髓肌肉萎縮症病友協會理事長、高醫大醫學院臨床醫學研究所講座教授鐘育志理事長補充,在新生兒眾多遺傳性疾病中,脊髓性肌肉萎縮症(SMA)為全世界嬰兒死亡率最高的遺傳疾病,每10,000-17,181位新生兒中就約有1位會患病。調查顯示,家長不進行加選SMA篩檢的三大主因為:
-
- 不懂篩檢原因:四成因為沒有SMA相關家族病史、三成七因為不理解「新生兒篩檢加選SMA項目」內容而不進行篩檢。
- 不懂嚴重程度:八成不知道SMA是世界上嬰兒死亡率最高的遺傳疾病。
- 不懂有藥可醫:七成八不知道SMA確診後有藥物可治療。
鐘育志理事長說,SMA屬於體染色體隱性遺傳疾病,六成患者會在六個月內發病,因負責製造運動神經元存活蛋白(SMN)的基因SMN1發生突變,使脊髓的運動神經元漸進性退化,產生肌肉無力、萎縮及行動困難等症狀。現在SMA不再是無藥可治的疾病,若能及早篩檢、發現與治療,即有更好的預後。目前台灣有三種治療藥物已通過藥證。一種為脊髓腔內注射藥物,每四個月施打一次;一種為口服藥物,每日服用;以上兩種藥品機轉為能改變或修正SMN2基因的剪接,以增加完整的SMN蛋白。另一種基因治療,採用緩慢靜脈輸注一次性給予劑量,終身施打一次,機轉為將SMN1基因導入患者細胞內,代替缺陷基因的功能以生成完整的SMN蛋白。健保署今年將放寬SMA治療藥物給付範圍,期盼下一步能透過健保的幫助,讓不同族群的病患都能獲得急需的治療。