
先天性腎上腺增生
臺北榮民總醫院 兒童遺傳內分泌科 楊佳鳳 醫師
臺北榮民總醫院 內分泌新陳代謝科 林宏達 醫師
本文原文刊載於中華民國內分泌學會出版之「腎上腺疾病新知」
一、前言
腎上腺分髓質與皮質兩部份。髓質主要分泌應付緊急壓力有交感神經作用的腎上腺素(epinephrine)和正腎上腺素 (norepinephrine);皮質則分泌皮質醇 (cortisol)、醛固酮 (aldosterone)以及各種雄性荷爾蒙如脫氫表雄固酮 (dehydroepiandrosterone, DHEA),雄固烯二酮 (androstenedione)和睪固酮 (testosterone)等。皮質醇主要功能是應付壓力、升糖並與免疫功能有關。醛固酮則作用於腎臟遠端細尿管,增加鈉離子的再吸收,排泄鉀和氫離子,使血壓上升。膽固醇是所有腎上腺皮質荷爾蒙的前驅物質,經過一連串酵素的作用而生成各種皮質荷爾蒙。先天性腎上腺增生症 (Congenital adrenal hyperplasia, CAH)為一染色體隱性遺傳疾病,根據不同類型和不同程度的類固醇生成阻滯,患者會有皮質醇、醛固酮和雄性荷爾蒙異常的表現。先天性腎上腺皮質增生症包括下面幾種類型,其中超過百分之九十為類固醇21-羥化酶 (Steroid 21-hydroxylase, 21-OH) CYP21A2的基因變異而引起。
1. 21-羥化酶 (21-hydroxylase, 21-OH)
2.11β-羥化酶 (11β-hydroxylase, 11β-OH)
3.17α-羥化酶 (17 α-hydroxylase, 17-OH;also known as 17,20-lyase)
4. 第 2 型 3β-羥類固醇去氫酶 (3β-hydroxysteroid dehydrogenase type 2, 3β-HSD2)
5. 類固醇生成急性調控蛋白 (Steroidogenic acute regulatory protein, StAR)
6. P450 膽固醇側鏈裂解酶 (P450 cholesterol side-chain cleavage enzyme, SCC)
二、先天性腎上腺增生症,21-羥化酶 (21-OH)缺乏
最常見的先天性腎上腺增生症,為21-羥化酶 (21-OH)缺乏,當腎上腺因21-羥化酶缺乏,無法合成皮質醇及醛固酮時,前驅物質17-羥助孕酮 (17-hydroxyproges-terone, 17-OHP)會升高,也導致促腎上腺皮質素 (adrenocorticotropic hormone, ACTH)升高,促使腎上腺增生肥大,並且往雄性荷爾蒙方向製造,使身體產生過多的雄性荷爾蒙,造成女嬰會有半陰陽外生殖器,如陰蒂肥大;男嬰則可能出現陰囊色深的症狀,21-羥化酶 (21-OH)缺乏的先天性腎上腺增生症是使新生兒外陰性別不明的最常見原因。兩性均可能出現毛髮增生症狀輔以非特異的胃腸症狀,如:脫水、腹瀉、嘔吐、食慾不振來表現。典型的先天性腎上腺增生症分為「失鹽型」(salt-wasting type)和「單純雄性化型」(simple-virilizing type)。大約3/4的患者是屬於「失鹽型」,會發生高血鉀、低血鈉、脫水與代謝性酸中毒,通常會於出生兩週內發生,如果未能及時治療,極可能因急性腎上腺機能不全,腎上腺危象而死亡。
非典型 (non-classical or late-onset type)先天性腎上腺增生症,患者酵素活性缺乏較輕微,可能出現多毛、月經不順、長痘痘…等等的男性化(virilization)症狀,合併骨齡超前、性早熟。這些患者需定時追蹤血液中17-羥助孕酮、睪固酮的濃度,以作為治療的評估依據,更需密切注意患者整體的發展,包含第二性徵發育程度以及骨齡的評估,以免出現性早熟,造成患者成人身高不如預期。目前在身高及性早熟治療方面,合併性荷爾蒙抑制劑和生長激素,臨床上確實能在生長板密合前改善患者身高。
雖然台灣已於80年代起,全面實施先天性腎上腺增生症新生兒篩檢,仍可能遺漏非典型的先天性腎上腺增生症此類比較輕症病患。如果此類病人診斷時,生長板已密合,身高上臨床可能無法提供具體的幫助。非典型先天性腎上腺增生症的成人女性患者可能出現多毛、月經失調以及不孕的問題,有時容易被誤診為多囊性卵巢症候群,針對此類患者除了檢驗血中相關生化值的濃度及基因確診外,也需考慮相關治療:月經失調可使用口服荷爾蒙來維持正常周期;因雄性激素過高無法排卵而造成的不孕,則應使用促進排卵的相關治療。
篩檢17-羥助孕酮 (17-OHP)濃度升高的新生兒典型 (嚴重型)21-羥化酶 (21-OH)缺乏症,在許多國家都已實行多年,當病患因新生兒篩檢疑似為CAH患者,接著需安排基因篩檢及遺傳諮詢,此症為體染色體隱性遺傳 (autosomal recessive inheritance, AR)模式,最常見的原因為第六號染色體上的CYP21基因有缺陷。夫妻雙方均為帶因者,其子女則有25%的機率正常,25%的機率會罹病,50%的機率與父母一樣是帶因者。實驗室可檢測血中皮質醇、雄性素 (androgen)和促腎上腺皮質素以及17-羥助孕酮進行進一步確認。21-羥化酶 (21-OH)催化膽固醇之代謝反應僅在腎上腺皮質層進行,從血液或任何體液均無法偵測21-羥化酶 (21-OH)的活性,因此無法藉由常規的生化檢查檢測21-羥化酶 (21-OH)濃度,故改為偵測代謝過程中之中間物質17-羥助孕酮。新生兒篩檢經由扎血於血片,檢驗17-羥助孕酮濃度是否偏高進行初步檢查,但因許多因素會造成假性上升如早產兒…等,所以仍需經過檢測血中皮質醇、雄性素和促腎上腺皮質素 (ACTH)以及17-羥助孕酮和基因檢測才能確認。
各國家地區先天性腎上腺增生症發生率不同,最高的二個地區分別是阿拉斯加的Yupik Eskimos 地區的 1:282 ;法國 La Reunion 地區的 1:2,141。據台灣 2002-2006年統計,發生率為1/20,800,如以每年二十萬名新生兒統計算,每年應可能發現10個病例。
三、基因與突變的介紹
先天性腎上腺增生症 (CAH)包括下面幾種類型:21-羥化酶(21-OH)、11β-羥化酶(11β-OH)、17α-羥化酶(17-OH)、第2型3β-羥類固醇去氫酶(3β-HSD2)、類固醇生成急性調控蛋白 (StAR)、P450膽固醇側鏈裂解酶 (SCC)以及P450氧化還原酶 (P450 oxidoreductase, POR)。21-羥化酶缺乏是最常見造成先天性腎上腺增生的原因,已如前述。下文就以此類型來介紹。
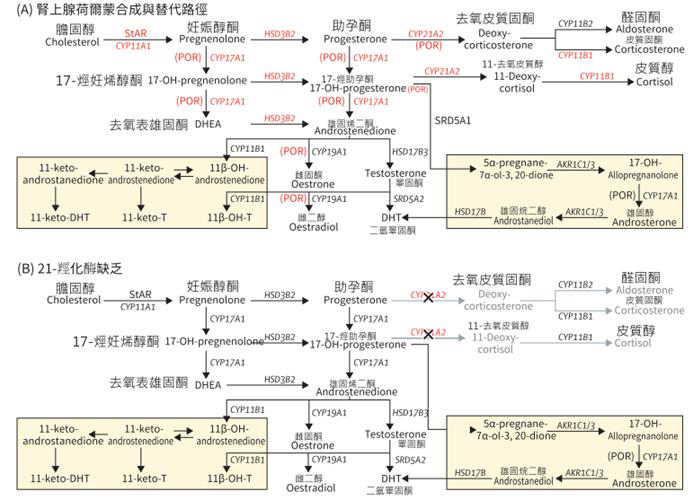 圖一、腎上腺類固醇生合成之路徑。
圖一、腎上腺類固醇生合成之路徑。
(A) 典型類固醇生成(steroidogenesis)路徑以及替代路徑 (淺黃底的部分)
(B) 21-羥化酶(21-OH)缺乏時的類固醇合成路徑。
(資料來源: El-Maouche D, Arlt W, Merke DP. Congenital Adrenal Hyperplasia. Lancet 2017;390:2194.)
21-羥化酶(21-OH)基因存在於第六對染色體之6p21.3處,分別與class III主要組織相容性複合體 (Human Histocompatibility Complex, MHC)之補體 (complement) C4基因交互並存,簡稱C4-CYP21 repeat module。21-羥化酶 (21-OH)分泌不足,和CYP21A2基因突變有關。此p21.3位置除含有補體C4基因,還有RP (STK29)、CYP21、TNX等基因。這些基因交互並存,排列成最常見的雙基因束 (RCCX bimodule),形成所謂 RP1-C4A-CYP21P-XA-RP2-C4B-CYP21-XB 之基因排列,或稱C4-CYP21 repeat module (R 表示RP、C 表示C4 與CYP21、X 表示XA )。在染色體p21.3處有二個P450c21基因存在;假性基因 (CYP21A1P, pseudogene)與活性基因 (CYP21A2, genuine gene)。假性基因序列有95%與CYP21A2基因相同。
在精、卵細胞行減數分裂(meiosis)的過程中,CYP21A2基因可產生交換 (crossover),並可能發生再重組 (recombination)。因此,引起CYP21A2基因變異的因素可分為三種:
1. 經由假性基因突變點小片段交換。
2. 自然突變 (spontaneous mutation)。
3. 混合基因型 (chimeric CYP21P/CYP21 與 TNXA/TNXB)。
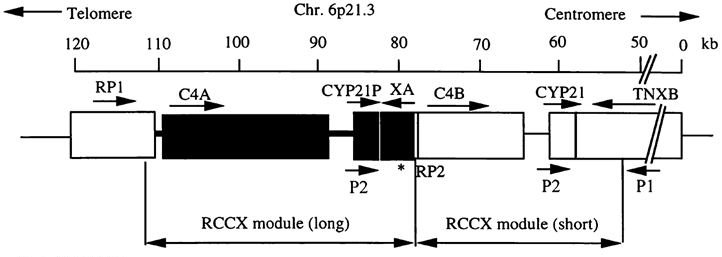 圖二、Bimodular form (RP1-C4A-CYP21P-XA-RP2-C4B-CYP21-TNXB) of the RCCX region of chromosome 6p21.3. (資料來源: Lee HH. Chimeric CYP21P/CYP21 and TNXA/TNXB genes in the RCCX module. Mol Genet Metab 2005; 84:4.)
圖二、Bimodular form (RP1-C4A-CYP21P-XA-RP2-C4B-CYP21-TNXB) of the RCCX region of chromosome 6p21.3. (資料來源: Lee HH. Chimeric CYP21P/CYP21 and TNXA/TNXB genes in the RCCX module. Mol Genet Metab 2005; 84:4.)
目前以次世代定序技術 (next generation sequencing, NGS)分析CYP21A2基因可分析所有的外顯子 (exon)及剪接位點 (splicing site)是否發生問題,但仍須經由傳統的桑格定序方法(Sanger sequencing)進行確認。
典型21-羥化酶 (21-OH)缺乏症的失鹽型和單純雄性化型分別反映出醛固酮缺乏的嚴重程度。失鹽型21-OH缺乏症,在沒有新生兒篩檢的情況下,極可能在出生2週內發生危及生命的腎上腺危象。單純雄性化型胎兒在媽媽肚子裡時,持續的暴露在過多的雄性素當中,使女嬰產生外生殖器男性化,此為最典型的症狀;而沒有早期診斷、早期治療的病患,因雄性素過多將導致性早熟症狀。非典型的21-OH缺乏症患者則保留高達50%的酵素活性,並且多數沒有明顯的腎上腺功能不全,但可能有部分糖皮質素缺乏症,並出現輕度雄性素過多的症狀。
其他先天性腎上腺增生症包括:11β-OH缺乏症、17-OH缺乏症(CYP17A1基因雖然只編碼一種酶,但該酶同時表達17α-羥化酶和17, 20-裂合酶活性)、3β -HSD2缺乏症、cytochrome P450 oxidoreductase缺乏症、類脂性先天性腎上腺增生 (lipoid congenital adrenal hyperplasia)、SCC 缺乏症 (P450 side-chain cleavage deficiency)。
四、治療
先天性腎上腺增生症的藥物治療包括使用糖皮質素 (glucocorticoid)以及鹽皮質素 (mineralocorticoid)進行;如有需要,也可於17-OH缺乏症、3β-HSD2缺乏症、類脂性先天性腎上腺增生、SCC缺乏症或POR缺乏症,這幾類患者身上使用性荷爾蒙補充療法。而性障礙患者之生殖器手術則為較具爭議之療法,先天性腎上腺增生症經常造成嬰幼兒性徵不明,然而是否要因性徵不明而進行性別重建或置換手術是一相當複雜的問題。也有研究指出46XX的先天性腎上腺增生症患者,可能因從胎兒時期就暴露在過多的雄性素之下,而有較高的Lesbian傾向。目前對於性別認同此一議題,專家學者傾向由個人心理認知決定,但因孩童時期醫療決策之決定權歸屬仍相當複雜,建議由相關領域專家,偕同病患及家屬共同討論決定。
除此之外,先天性腎上腺增生症的治療挑戰還包括如何避免皮質醇的過度治療,以及性激素失衡的控制。長期併發症包括異常生長發育、對骨骼和心血管系統的不良影響以及不孕症。以藥物試著平衡身體如此重要的激素治療,實為不易,臨床上需要相當之治療經驗和病患、家屬的長期配合,目標為減少皮質醇的過度暴露和長期補充的副作用,改善荷爾蒙控制,並模仿生理荷爾蒙模式。病患需長期觀察記錄生長速度並追蹤骨齡,如出現生長遲滯,身材矮小合併性早熟,臨床上顯示適當時機使用性荷爾蒙抑制劑合併生長激素,於生長板密合前可有效改善身高。
糖皮質素缺乏為先天性腎上腺增生症患者最嚴重的疾病生理問題,可能威脅生命。在瑞典的一項研究中,有588名先天性腎上腺增生症患者,病患死亡大多因腎上腺危象引起,突出了這種狀況的重要性。但患者長期接受糖皮質素治療則可能引起身材矮小,因此糖皮質素補充,應該以盡可能低的有效劑量為目標。
先天性腎上腺增生症患者目前治療效果無法十分完美,可能原因為荷爾蒙失調或與治療相關的合併症,因此開發新療法的病理生理學仍在持續進行中。其中,口服緩釋氫皮質酮製劑 (modified-release oral hydrocortisone)成功降低了患者體內雄性荷爾蒙的濃度;藉用連續胰島素幫浦,模擬晝夜節律,規律地皮下注射氫皮質酮,顯示出對ACTH的充分抑制以及與常規治療相比較低的皮質酮總劑量。先天性腎上腺增生症是一種單基因疾病,因此,基於細胞和基因編輯的基因治療技術已經在腎上腺皮質機能不足的動物模型中獲得成功,未來的技術和基因進步可能治癒先天性腎上腺增生病患。
五、總結
台灣新生兒篩檢,已於民國80年代將先天性腎上腺增生症 (CAH)加入必須篩檢項目,目前台灣出生之新生兒幾乎為百分之百受檢。台灣新生兒篩檢之效率、涵蓋率和確診後治療成效,不論是先天性腎上腺增生症或其他疾病,皆不亞於甚至勝過其他先進國家。
目前基因診斷、基因治療發展迅速,在基因診斷方面,以目前全外顯子定序 (WES)/全基因體定序 (WGS)的效率,即使是診斷涵蓋所有其他少見類型的先天性腎上腺增生症也不需花費很長的時間,基因診斷的技術不斷改良,我們期待能夠透過早期診斷、早期治療,盡量減低先天性腎上腺增生症對患者造成的影響。
WES/WGS將全面性協助許多可治療疾病之早期診斷,在新生兒方面可為需治療或追蹤之疾病提供更快速的診斷;而基因治療將改變許多遺傳疾病的治療模式,我們正站在許多疾病診斷和治療模式劇烈改變的風口,未來一定能提供病患更佳的預後。
誌謝
感謝蘇郁文醫師的建議
參考文獻
1. Bidet M, Bellanne-Chantelot C, Galand-Portier MB, et al. Fertility in women with non- classical con genital ad re nal hyperplasia due to 21-hydroxylase deficie ncy. J Clin En do- crinol Metab 2010;95:1182-1190.
2. El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet 2017;390:2194- 2210.
3. Lee HH. Chimeric CYP21P/CYP21 and TNXA/TNXB genes in the RCCX module. Mol Genet Metab2005;84:4-&
4. Bidet M, Bellanne-Chantelot C, Galand-Portier MB, et al. Clinical and molecular charac- terizati on of a cohort of 161 un related wome n with non classical congen ital adre nal hy- perplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab 2009;94:1570-1578.
5. Fisher AD, Ristori J, Fanni E, Castellini G, Forti G, Maggi M. Gender identity, gender as- signment and reassignment in individuals with disorders of sex development: a major of dilemma. J Endocrinol Invest 2016;39:1207-1224.
6. Falhammar H, Frisen L, Norrby C, et al. Increased mortality in patients with congen- ital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2014;99:E2715-2721.
7. White PC. Neonatal screening for congenital ad renal hyperplasia. Nat Rev Endocrinol 2009;5:490-49&
8. Mallappa A, Sinaii N, Kumar P, et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adre- nal hyperplasia. J Clin Endocrinol Metab 2015;100:1137-1145.
9. Bryan SM, Honour JW, Hindmarsh PC. Management of altered hydrocortisone pharma- coki netics in a boy with con genital ad re nal hyperplasia using a continuous subcuta ne- ous hydrocortisone infusion. J Clin Endocrinol Metab 2009;94:3477-3480.
10. Nella AA, Mallappa A, Perritt AF, et al. A phase 2 study of continuous subcutaneous hy- drocortis one infusion in adults with congenital ad renal hyperplasia. J Clin Endocrinol Metab 2016;101:4690-4698.
11. Balyura M, Gelfgat E, Ehrhart-Bor nstein M, et al. Tran spla ntati on of bovine ad re no corti- cal cells en capsulated in algin ate. Proc Natl Acad Sci U S A 2015;112:2527-2532.